5.0 Create a Nuclease Manifest¶
The nuclease manifest stores information about each nuclease mechanism used to introduce double-stranded breaks (DSBs). Broken String Biosciences provides an empty template manifest to ensure all required columns are included.
Note
Do not modify the template directly. Always duplicate the template schema first (see steps below).
Create a nuclease manifest from the template¶
- Select Data Registry from the left sidebar.
- Hover over Nuclease Manifest Template under the Templates v2.0 project.
- Select the ellipsis (
...), then select Duplicate Table Schema. - Enter a name for the new nuclease manifest.
- Under Destination Project, select the registry project to add it to (see Create a Registry).
- Select Create.
You can now populate the manifest by:
- Adding nuclease data manually — recommended when using a limited number of nuclease mechanisms.
- Importing a manifest CSV file — useful for batch entry in external software such as Microsoft Excel.
Nuclease type defaults¶
Cas9¶
When nuclease_type is set to cas9, the following defaults apply:
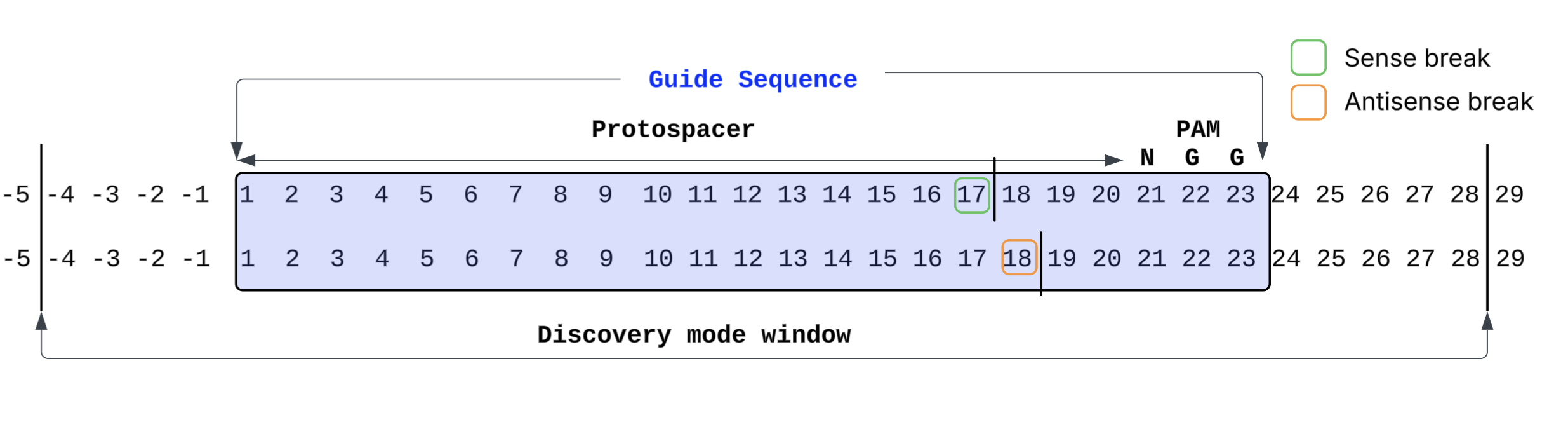
- PAM:
NGGat the 3' end of 20 bases of protospacer guide sequence - Merge distance:
0(only adjacent breaks are merged) - Expected cut site: blunt cut between bases 17 and 18 relative to the start of the guide, 3 bases upstream of the PAM start
Cas12a¶
When nuclease_type is set to cas12a, the following defaults apply:
- PAM:
TTTVat the 5' end of 22 bases of protospacer guide sequence - Merge distance:
4(breaks within 4 bases are merged) - Expected cut site: staggered cut
- Sense break between bases 22 and 23 relative to guide start (including PAM), 18 bases downstream of PAM end
- Antisense break between bases 27 and 28 relative to guide start (including PAM), 23 bases downstream of PAM end
Note
Default guide length and PAM values are overridden by the guide_sequence and
pam values you enter in the manifest.
Custom¶
When nuclease_type is set to custom, no defaults are applied. The guide_sequence
and pam fields are mandatory.
Leaving the sense_break and antisense_break fields blank runs the pipeline in
discovery mode: any break within a guide-like sequence in the genome, plus or minus
5 bases, is counted as homology-based. After reviewing the discovery mode outputs,
you can re-run with specific sense and antisense break values derived from empirical
observation.
The diagram below shows the guide sequence position numbering and discovery mode window: